What is the physical significance of the oscillator strength? Following Werner Kuhn's arguments (e.g. in
this paper), it marks the number of electrons oscillating per spatial dimension during an electronic transition. The sum over the oscillator strengths of all the excited states amounts to the number electrons, which is the essence of the
Thomas-Reiche-Kuhn sum rule. In other words, the oscillator strength counts how much of the total oscillating potential is used for a specific transition.
This interpretation explains for example the linear relationship of the oscillator strength of the lowest excited state with system size in the case of some conjugated organic polymers (see e.g.
this paper): If there are more electrons available to oscillate, then the transition strength increases.
The
oscillator strength f
ij between two non-degenerate states i and j is defined (in atomic units) as two thirds of the squared transition dipole moment multiplied by the energy gap
where the vector
r contains all 3N spatial coordinates of the N electrons
The Thomas-Reiche-Kuhn sum rule now states that the sum over the oscillator strengths from one state i to all possible other states is equal to the number of electrons in the system, i.e.
In particular, if we consider excitations from the ground state, then all oscillator strengths are positive. Which means that the oscillator strengths can in fact be viewed as a partitioning of the number of electrons.
The derivation of this sum rule starts by realizing that the momentum operator with respect to any spatial coordinate x of any particle (e.g. x=y
2) is given as the commutator of the Hamiltonian with this coordinate
This follows whenever H is of the form
where clearly the derivatives with respect to x are the only part, which does not commute with x itself
By applying the product rule twice, the first term of this expression becomes
And in summary
The remaining proof follows what is shown
here (sorry that I am switching the notation, but I copy-and-pasted a little bit ...). First one realizes that the commutator of x and p
x is equal to i
Then one expands the commutators and inserts a resolution of the identity over the eigenstates of the Hamiltonian
Insert the above expression for p
x
The commutators are evaluated by letting H act either on the bra or the ket, which results in a multiplication with the respective eigenvalue. And after summing together the equivalent terms one obtains
The actual
r vector was composed of 3N individual electron coordinates. The above equation holds for each of these coordinates. Thus, in summary:
which is just what we wanted to show.
Edit: The discussion is also related to the relation of the length and velocity gauge for computing oscillator strengths. Some more information here.




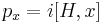
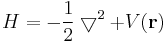
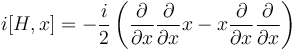
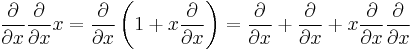

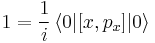


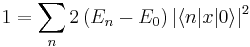









 After excitation (γ means "photon" [2]) the cis-doublebond becomes a single bond. Because of that it stretches out. Now there is an ecliptical single bond with weak π-conjugation. It spontaniously rotates.
After excitation (γ means "photon" [2]) the cis-doublebond becomes a single bond. Because of that it stretches out. Now there is an ecliptical single bond with weak π-conjugation. It spontaniously rotates.



